

Dr. Brenda Canine
Research Interests:
Diabetes (Misfolded Insulin protein)
Alzheimer’s Disease (Misfolded Tau protein)
Mental Health in Rural America
Assistant Professor McLaughlin Research Institute Associate Course Director for Microbiology Touro College of Osteopathic Medicine - Montana
Education
University of Idaho, Moscow, ID; BS, Chemical Engineering, 2010
Washington State University, Pullman, WA; PhD, Pharmaceutical Sciences, 2010
McLaughlin Research Institute, Postdoctoral Fellow, 2013

We study misfolded proteins to find improved treaments for Diabetes, Neurodegeneration and Mental Health.
-Brenda Canine

-
2023-Present; Assistant Professor, Associate Course Director of Microbiology
2017- 2023; Tenured Biology Faculty- Great Falls College MSU
2018-Present; Content Creator/Expert Carolina Biological
2013-PresentAffiliate Faculty McLaughlin Research Institute
2018-22; MSGC Campus Affiliate Member Contact for GFC MSU
2018-21; Bridges to Baccalaureate NIH Grant with Montana State University
2016; Montana Educator of the Year Award- Montana Agate
2015; Department of Labor Montana HealthCARE grant -TAACCCT
2016 – 2017; Instructional Designer - Great Falls College MSU
2013 – 2017; NANSLO Lab Manger - Great Falls College MSU
2012 – 2017; Adjunct Instructor – Great Falls College MSU
2014; Montana Space Grant Consortium Educational Enhancement Grant
2014; Great Falls College MSU Innovation Award
2012-16; Department of Labor Consortium for Healthcare Education Online Grant-TAACCCT
2011 – 2011; Postdoctoral Researcher --McLaughlin Research Institute
2008 – 2010; Graduate Student Teaching Assistant: Washington State University, College of Pharmacy
2009; Graduate Student Intern: Gilead Sciences, Foster City (San Francisco) CA
2005 – 2006; University of Idaho and Northern Arizona University, Moscow, ID Engineering Senior Design Project Lead
2015; GFC MSU Dean’s Award for Innovation
2013; Best Postdoc presentation PRION International Conference
2010-11; AFPE Pre-doctoral Fellowship
2006-08; Graduate Student Scholar Award, WSU
-
I joined the founding faculty of the first medical school in Montana in May of 2023. As an Assistant Professor at Touro University College of Medicine Montana, I will study disease mechanisms and educate future scientists. Prior to accepting my current position, I was Biology Faculty at Great Falls College MSU where I received tenure in Fall 2022. This was a full-time teaching appointment and taught Microbiology Human Biology, Anatomy and Physiology, Pathophysiology, and Chemistry.
As an instructor, I can identify students interested in pursuing biomedical research and help guide them to opportunities in that area. I have worked in a variety of research settings and can help students by answering questions and assisting them in the application process. I had fantastic mentors as an undergraduate student and credit those faculty members who took extra time to help develop my interests with helping shape my current career. As a student who was a woman in a male dominated field (Engineering) I often found myself singled out in courses and I can bring that perspective to students who also may also feel dissimilar to their peer group. I am passionate about providing science opportunities to students and see my role as an educator as one who helps make academic and opportunity connections. I am the campus contact for the Montana Space Grant Foundation and have assisted with multiple student projects in the last few years. I am also a board member of the Dr. David Baker Memorial Student Science Foundation (DWBF), which works to help provide opportunities and monetary support to K-12 students in Montana pursuing STEM projects. I have been helping with the Montana Region II Science Fair since 2012 and have mentored many student projects for high school and college students.
Prior to becoming a full-time faculty member, I was an Instructional Designer and remote science lab manager. I helped design and create lab activities for the North American Network of Science Labs Online (NANSO), an international network of remote web-based science labs which allowed students to remotely control lab equipment in real time over the Internet to complete science labs for online courses. I earned a Bachelor of Chemical Engineering degree from the University of Idaho and a doctoral degree in Pharmaceutical Sciences from Washington State University and then completed a postdoc in Genetics and Systems Biology of Neurodegenerative Disease at the McLaughlin Research Institute. I have worked in government labs including the Idaho National Laboratory and the Pacific Northwest National Laboratory, academic labs at Washington State University, University of Idaho, Arizona State University and in industry at Gilead Sciences.
I currently bring an interdisciplinary background to science instruction in the biological sciences at Great Falls College MSU. My research background is unusual at a 2-year college level and has allowed me to work with students on a wide range of projects. I have been a Montana Space Grant Consortium affiliate representative and helped students complete their projects all with small budgets and scopes of work appropriate for introductory undergraduate students.
Finally, in other work, I built and developed a remote web-based science lab for Great Falls College MSU from 2013-2017. This work was funded by a Department of Labor Workforce Development grant and allowed us to have students at remote distances log in and complete lab activities using lab equipment in real time. Students from Alaska, Colorado, and Montana utilized this lab. We also did proof of concept demonstrations with faculty members from across the world including locations in the Northern Mariana Islands, Hawaii, Texas, and California. The Great Falls, MT, lab was one of three labs in a network of labs called the North American Network of Science Labs online. In addition to offering labs, this project resulted in OER curriculum development for online lab courses.
-
My postdoctoral research work utilized mouse genetics combined with systems biology approaches to understand neurodegenerative disease pathways in prion disease, frontotemporal dementia and other neurodegenerative diseases where protein accumulation is a pathological finding. This work lead to a number of key publications that define fundamental aspects of neurodegeneration including core transcriptional regulatory circuits, mouse genotype-specific differences in protein expression in neurodegeneration, pre-clinical events that might predict accumulation of toxic proteins.
Kim, TK, Lee, I, Cho, JH. Canine, B. Keller, A. Price, N. Hwang, D. Carlson GA, Hood L. Core transcriptional regulatory circuits in prion disease. 2020 Mol Brain 13:10
Gunn, TM, Canine, B. Application of mouse genetics to human disease: Generation and analysis of mouse models. Chapter 5 in Rosenburg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease. 2014 Nov; Elsevier Press, Amsterdam.
Orr ME, Pitstick R, Canine B, Ashe KH, Carlson GA. Genotype-specific differences between mouse CNS stem cell lines expressing frontotemporal dementia mutant or wild type human tau. PLoS One. 2012; 7(6) e39328.
Westaway D, Genovesi S, Daude N, Brown R, Lau A, Lee I, Mays CE,Coomaraswamy J, Canine B, Pitstick R, Herbst A, Yang J, Ko KW, Schmitt-Ulms G, Dearmond SJ, McKenzie D, Hood L, Carlson G Down regulation of Shadoo in prion infections traces a pre-clinical event inversely related to PrP accumulation. PLos Pathog, 2011 Nov; 7(11): e1002391.Epub Nov 2011.
My graduate research utilized cell biology and protein engineering for the development of novel protein polymers to design drug delivery vehicles for gene therapy applications specifically for cancer cell treatments. Cationic protein polymers were designed, produced, characterized and evaluated for their efficacy in cancer cell models specifically ovarian cancer cell lines.
Canine B, Wang Y, Ouyang W, Hatefi A. Development of targeted recombinant polymers that can deliver siRNA to the cytoplasm and plasmid DNA to the cell nucleus. J Control Release. 2011, 151(1): 95-101.
Canine BF, Hatefi A. Development of recombinant cationic polymers for gene therapy research. Adv Drug Deliv Rev. 2010;62(15):1524-1529.
Canine B, Wang Y, Hatefi A. Biosynthesis and characterization of a novel genetically engineered polymer for targeted gene transfer to cancer cells. J Control Release. 2009, 138(3): 188-96.
Canine B, Wang Y, Hatefi A. Evaluation of the effect of vector architecture on DNA condensation and gene transfer efficiency. J Control Release. 2008, 129(2):117-23.
Other collaborative contributions include research on gene targeting with a focus on vector design strategies, in particular. This research focused on targeting cancer via iNOS gene therapy, cancer suicide gene therapy, and development of biomimetic vectors generally focused on breast cancer cells.
McCarthy H, Zhobbenko Y, Wang Y, Canine B, Robson T, Hirst D, Hatefi A. Evaluation of a multi-functional nanocarrier for targeted breast cancer iNOS gene therapy. Int J Pharm. 2011, 405(1-2): 196-202.
Wang Y, Canine B, Hatefi A. HSV-TK/GCV cancer suicide gene therapy by a designed recombinant multifunctional vector. Nanomedicine. 2011, 7(2):193-200.
Sastry MSR, Canine B, Wang Y, Hatefi A. Development of a genetically engineered biomimetic vector for targeted gene transfer to breast cancer cells. 2009, Mol Pharm 6(4): 1100-1109.
Wang Y, Sastry MSR, Canine B, Hatefi A. A designer biometric vector with a chimeric architecture for targeted gene transfer. J. Control Release. 2009, 137(1):46-53.
-

Misfolded Proteins
Proteins are the catalysts of chemical reations in our cells. Often described as a lock and key mechanism, changes ot the structure of the lock or the key will result in the catalyst not working optimally. Dysfunction in protein structure is a known cause of many diseases from Cystic Fibrosis to Sickle Cell Anemia to Muscular Dystrophy. This lab studies misfolded proteins in their roles in chronic diseases associated with aging.
-
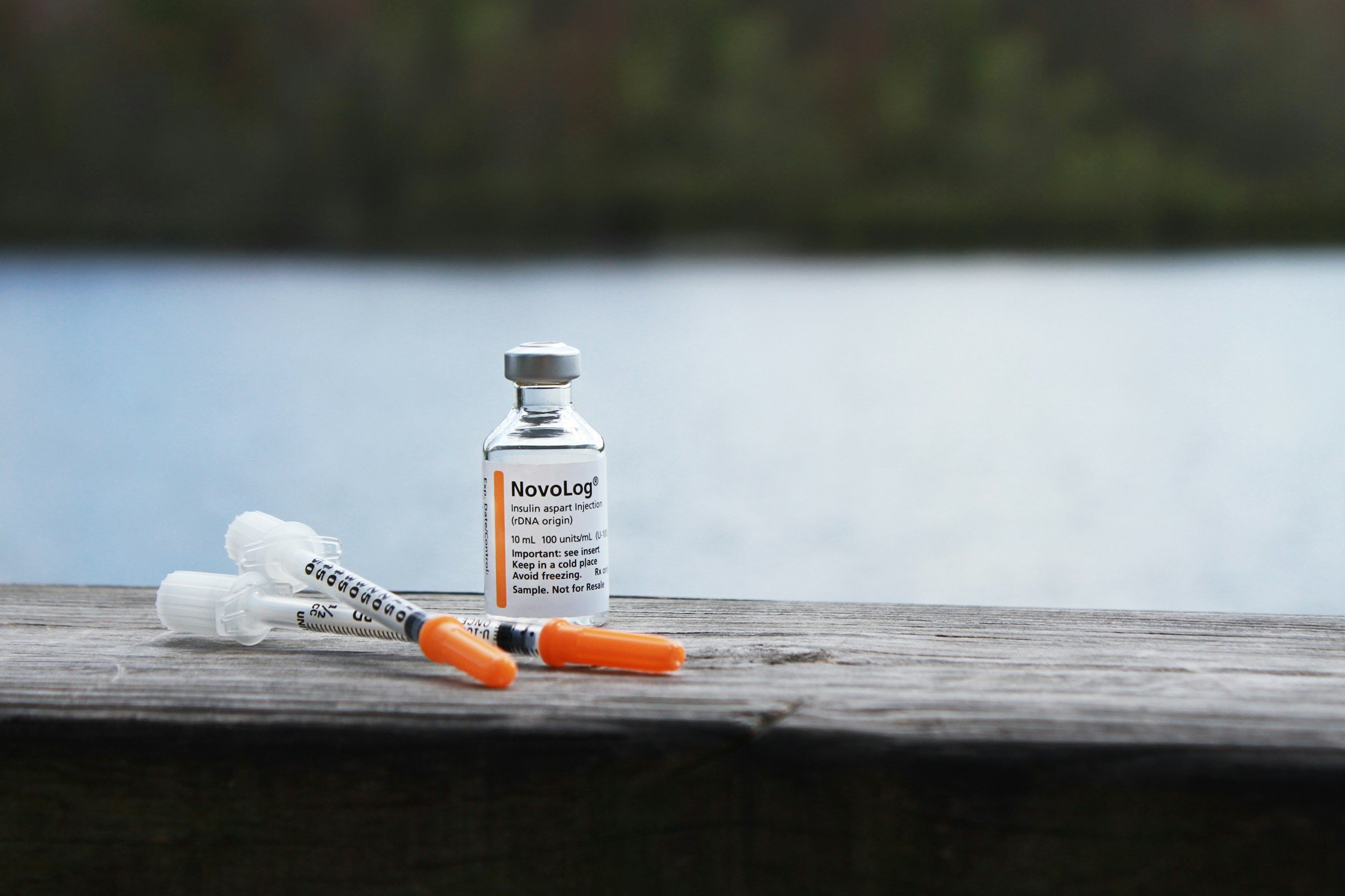
Diabetes
Insulin is an important protein hormone that is involved in systemic glucose utilization to maintain homeostasis. In both Type 1 and Type 2 Diabetes, insulin signaling is impaired. Treatment in Type 1 involves exogenous insulin delivery through subcutaneous injection or infusion and Type 2 beings with oral medications with various mechanisms of action, however 40% of patients fail oral medication therapy and progress to requiring insulin supplementation to maintain glycemic control.
Large boluses of insulin delivered subcutaneously can result in a condition called insulin-derived amyloidosis where the aggregated amyloid mass positively stains with anti-insulin antibodies. Additionally, during insulin infusion therapy, it has been repeatedly reported that occlusions can occur due to aggregated insulin in the pump tubing and cartridges. We strive to understand how insulin misfolding may lead to aggregation adn amyloid foramation.
.
-

Neurodegeneration
Prion diseases belong to group of progressive conditions that affect the nervous system in humans and animals. In people, prion diseases impair brain function, causing memory changes, personality changes, a decline in intellectual function (dementia), and problems with movement that worsen over time. The signs and symptoms of these conditions typically begin in adulthood, and these disorders lead to death within a few months to several years.Familial prion diseases of humans include classic Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and Fatal Familial Insomnia (FFI). These conditions form a spectrum of diseases with overlapping signs and symptoms.
-

Mental Health
In the last decaten Montana has consistently placed 1st, 2nd or 3rd for the most suicdes nationally. As a rural state in the shadows of the Rocky Mountains what factors contribute to this high number. Working with the Montana Department of Health and Human Services Dr. Canine, fellow faculty and studets at Touro University Collehe of Medicine in Great Falls are hopng to understand the risk factors unique to Montana.
Prion Diseases- the first group of diseases shown to be transmissible by misfolded proteins
-
What are Prion Diseases?
Prion diseases belong to group of progressive conditions that affect the nervous system in humans and animals. In people, prion diseases impair brain function, causing memory changes, personality changes, a decline in intellectual function (dementia), and problems with movement that worsen over time. The signs and symptoms of these conditions typically begin in adulthood, and these disorders lead to death within a few months to several years.Familial prion diseases of humans include classic Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and Fatal Familial Insomnia (FFI). These conditions form a spectrum of diseases with overlapping signs and symptoms.
-
How common are Prion Diseases?
These disorders are very rare. They affect about one person per million worldwide each year. Approximately 300 cases occur annually in the United States.
-
What genes are related to prion diseases?
Mutations in the PRNP gene cause prion disease. Only a small percentage of prion disease cases run in families. Most cases are sporadic, which means they occur in people without any known risk factors or gene mutations. Rarely, prion diseases can be transmitted by accidental exposure to prion-contaminated tissues during a medical procedure. This type of prion disease is called iatrogenic.One type of prion disease in humans, variant Creutzfeldt-Jakob disease (vCJD), is acquired by eating beef products obtained from cattle with prion disease. In cows, this form of the disease is known as bovine spongiform encephalopathy (BSE) or, more commonly, "mad cow" disease. Another example of an acquired human prion disease is kuru, which was identified in the South Fore tribe in Papua New Guinea. The disorder was transmitted when tribe members ate the tissue of affected people during cannibalistic funeral rituals.Familial forms of prion disease are caused by inherited mutations in the PRNP gene. This gene provides instructions for making a protein called prion protein (PrP). Normally, this protein is likely involved in transporting copper into cells. It may also play a role in protecting brain cells and helping them communicate. In familial cases of prion disease, mutations inthe PRNP gene cause cells to produce an abnormal form of the prion protein known as PrPSc. In iatrogenic and acquired cases, an affected person develops prion disease from exposure to this abnormal protein.In a process that is not fully understood, PrPSc has the ability to convert the normal prion protein, PrPC, into more PrPSc. This abnormal protein builds up in the brain, forming clumps that damage or destroy nerve cells. The loss of these cells creates microscopic sponge-like holes in the brain, which leads to the signs and symptoms of prion disease.
-

-
Anti-prion ASOs prolong survival in mice with prion disease
From: Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints
Eric Vallabh Minikel et al., 2019 Nucleic Acids Research, 2020, Vol. 48, No. 19 10615–10631
-
